- 结果文件
- 使用指南
- 版本记录
- 联系客服
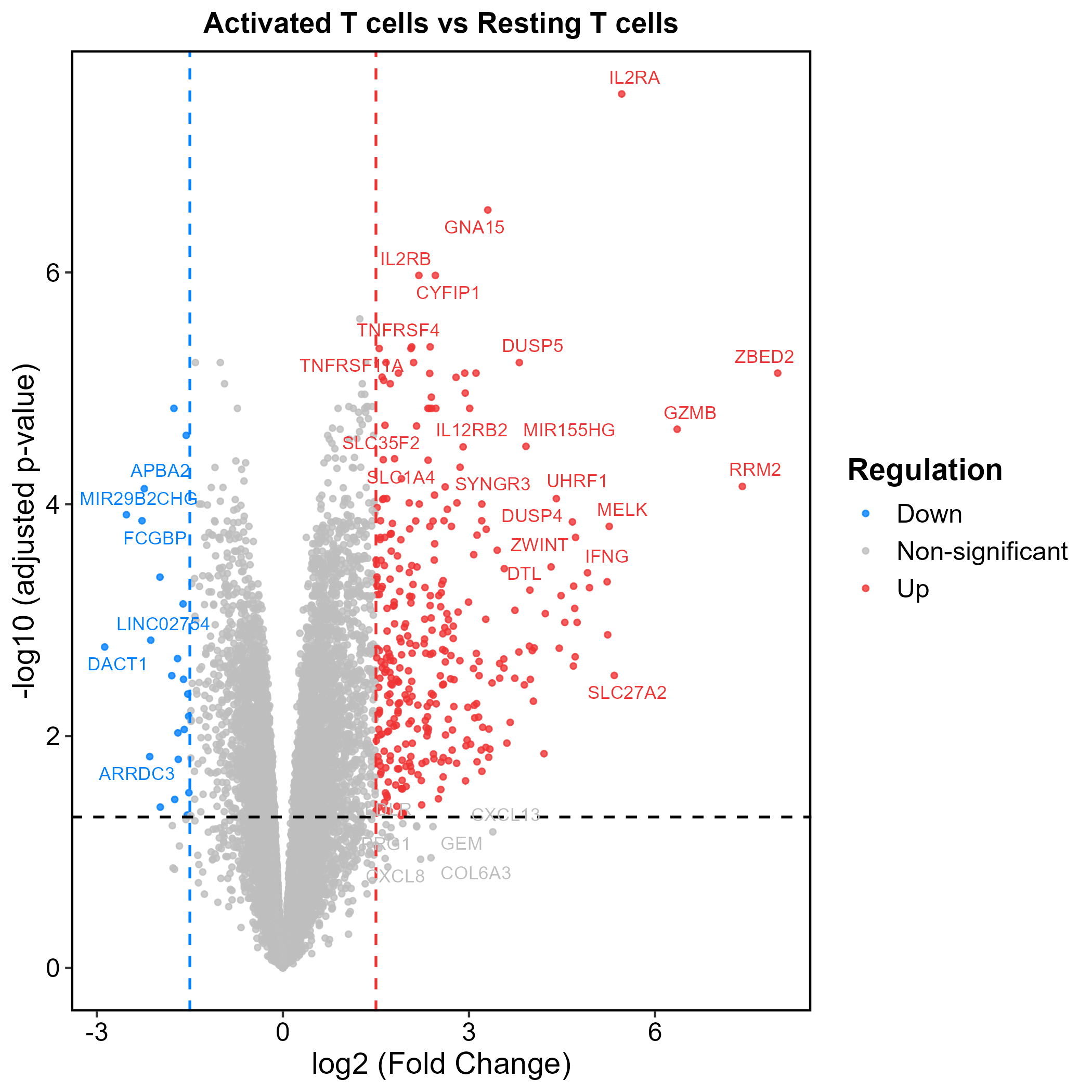
差异分析(Differential Analysis),转录组等组学研究中最基础且核心的分析内容之一,以生物学意义的方式定性基因表达量,计算两个比较组的基因表达差异倍数(Fold change),得到显著性P值,然后通过多重假设检验矫正筛选出具有统计学意义的显著差异表达基因,从而为我们挑选目标基因缩小范围,再将结果与后续分析点相结合,如富集分析等,以探究相关的生物学过程和分子机制。
我们一般会使用limma/DESeq2进行差异分析,再将结果使用火山图等图表进行可视化。
DESeq2 是为 RNA-seq 设计的,RNA-seq 数据是离散的“计数”(Counts),例如某个基因被测序仪读到了 1000 次。这种数据通常具有“均值-方差关系”(Mean-Variance Relationship),即表达量高的基因方差也大。DESeq2 使用广义线性模型(GLM),并假设数据服从负二项分布,这能更好地捕捉计数数据的离散特性。适用于RNA-seq 的原始计数(Raw Counts);
Limma 通常使用 TMM (Trimmed Mean of M-values) 或 Quantile Normalization。TMM 假设大部分基因没有差异表达,通过调整样本间的缩放因子来校正技术偏差,当处理微阵列数据,或者已经将 RNA-seq 的原始计数转换成了logCPM (log-counts per million) 或 TPM/FPKM 等连续值,且数据分布接近正态时推荐使用Limma。
输入文件:
① 基因表达矩阵,每一行代表一个基因,每一列代表基因在个样本中的定量结果(整数)

② 差异比较组,第一列为样本ID,第二列为差异比较组(两组样本间比较)
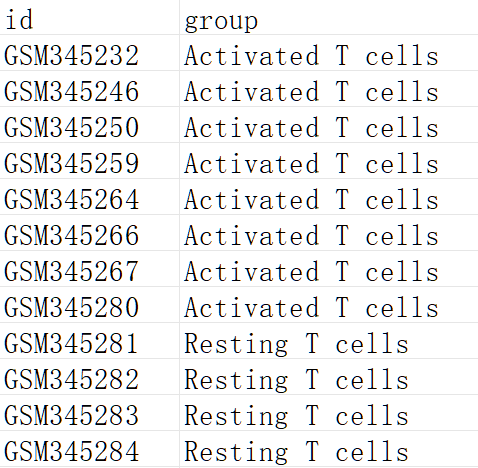
支持制表符分隔( .tsv、.txt) 和 逗号分隔(.csv)文件
小工具结果:
deg_res_file.csv
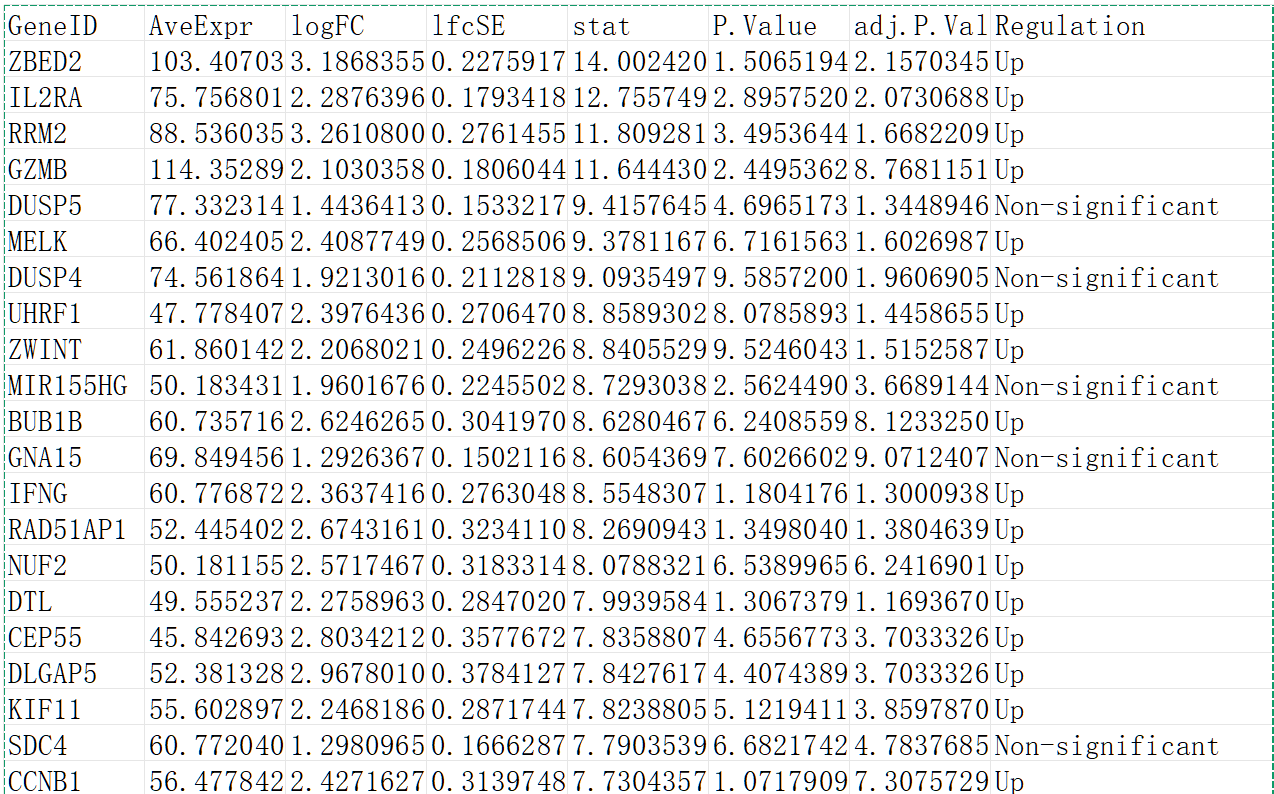
Gene ID:基因标识符;AveExpr:平均表达量,该基因在所有样本中的平均表达水平;logFC:对数倍数变化,实验组相对于对照组的表达量变化倍数(取log2),正数(上调,Up-regulated),负数(下调,Down-regulated);lfcSE:标准误,衡量 logFC 估计值的波动范围,越小说明估计越稳定;stat:t 统计量,用于计算 P 值的统计量,值越大(绝对值)通常差异越显著;P.Value:原始 P 值,差异表达的显著性水平,越小越显著(通常 < 0.05 认为显著);adj.P.Val:校正 P 值,经过多重检验校正(如 FDR)后的 P 值;Regulation:调控状态,根据 logFC 和显著性判断的结果(Up,显著上调 | Down,显著下调 | Non-significant,无显著差异)
plot.pdf&plot.png