- 结果文件
- 使用指南
- 版本记录
- 联系客服
本工具用于基于BLAST进行核酸序列多序列比对,可选择不同的比对算法以适应不同使用场景,如使用blastn-short算法进行引物特异性评估
比对模式说明:
| 高速、高严格度 | 比对高度相似(≥95% 同源性)的长序列,例如: | 使用长种子序列(28bp)的贪婪算法。要求连续匹配的种子区域才能启动延伸,因此速度极快,但对错配(差异)非常敏感。 | ⚡⚡⚡ |
| 中等速度与灵敏度 | 比对中度相似的序列,例如: | 使用不连续种子(如模式 | ⚡⚡ |
| 高灵敏度、低速度 | 寻找低相似度的序列,例如: | 使用短种子(11bp)进行搜索。种子短,更容易找到匹配点,因此灵敏度最高,但也会导致速度最慢,并可能产生更多无关的随机匹配,需仔细评估E值。 | ⚡ |
| 为短序列优化 | 专门用于短序列查询(长度 ≤ 50 bp),例如: | 调整了打分矩阵和参数设置,优化了短序列的比对能力,避免因其长度短、错配容忍度低而被过滤掉。查询序列长于30bp时,效果开始下降。 | ⚡⚡ |
输入文件:
查询序列(Query Sequence):核酸序列文件,FASTA格式,待分析的序列,当进行短序列查询(如:引物特异性评估),要求每条序列长度 <50bp ,比对模式选择 blastn-short 。

参考序列(Reference Sequence,用于建库):参考序列是用于构建BLAST数据库的已知序列集合。这些序列通常来自公共数据库(如NCBI的GenBank、UniProt等)或用户自定义的序列集(如全基因组序列),BLAST会将查询序列与这些参考序列进行比对。

小工具结果:
blast_results.xls
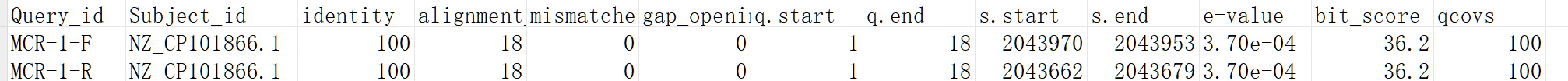
query id:查询序列ID标识;refer id:参考序列ID标识;identity (%):序列比对的一致性百分比;alignment length:符合比对的比对区域的长度;mismatches:比对区域的错配数;gap openings:比对区域的gap数目;q.start:比对区域在查询序列上(query id)的起始位点;q.end:比对区域在查询序列上(query id)的终止位点;s.start:比对区域在参考序列上(refer id)的起始位点;s.end:比对区域在参考序列上(refer id)的终止位点;e-value:比对结果的期望值,将比对序列随机打乱重新组合,与数据库进行比对,如果功能越保守,则该值越低;bit score:比对结果的bit score值;qcovs:查询的覆盖率。
当前版本为1.0版本,上架时间为:2025-09-05