- 软件介绍
- 软件与数据库
- 使用指南
- 版本记录
- 相关课程推荐
本流程用于细菌基因组纯二代PE文库测序在线自动化、批量组装、注释和统计。适用于如下两种类型:
①从原始数据开始,包含组装和注释;
②从组装结果开始,包括各类注释分析。

基因组组装:SPAdes+SOAPdenovo
CDS预测:Glimmer(http://ccb.jhu.edu/software/glimmer/index.shtml,V3.02)
tRNA预测:tRNAscan-SE(http://trna.ucsc.edu/software/)
rRNA预测:Barrnap(https://github.com/tseemann/barrnap/)
基因功能注释:
比对软件:DIOMAND(V0.9.24.125,http://github.com/bbuchfink/diamond)
比对数据库:
NR:NCBI创建并维护的非冗余蛋白数据库,数据库内容较为全面,可作为基因注释结果的主要参考。
Swiss-Prot(https://web.expasy.org/docs/swiss-prot_guideline.html):由欧洲生物信息学研究所(European Bioinformatics Institute,EBI)维护的非冗余基因数据库,注释结果大多经过实验验证,可靠性较高,但收录基因数量很少。
Pfam(http://pfam.xfam.org/):依赖于由多序列比对和隐马尔可夫模型(HMMs),可给出基因中包含的所有结构域信息。
COG(http://www.ncbi.nlm.nih.gov/COG/):Clusters of Orthologous Groups of Proteins的缩写,可以对预测蛋白进行功能注释、归类以及蛋白进化分析。构成一个COG对应于一个古老的保守结构域,每个COG的蛋白被假定来自于同一个祖先蛋白。
GO(http://www.geneontology.org/):基因本体论Gene Ontology的缩写,为标准化不同物种、不同数据库的生物学术语而创立的数据库,可获得标准化的功能描述信息。
KEGG(http://www.genome.jp/kegg/):是系统分析基因功能,联系基因组和功能信息的大型知识库,其丰富的通路信息有助于从系统水平去了解基因的生物学功能,例如代谢途径、遗传信息传递以及细胞学过程等一些复杂的生物过程。
基因组圈图绘制:Circos Version 0.69-6(http://www.circos.ca)
特定数据库注释:
基因组岛:islandpath
前噬菌体:PhiSpy
CRISPR-Cas系统:CRISPRFinder
碳水化合物活性酶:CAZy(http://www.cazy.org/)
毒力基因:VFDB
耐药基因:CARD&ResFinder
病原菌与宿主互作:PHI(http://www.phi-base.org/)
跨膜蛋白:TMHMM
转运蛋白:TCDB
分泌蛋白:SignalP(http://www.cbs.dtu.dk/services/SignalP)
次级代谢产物合成基因簇:antiSMASH Version 8.0
细菌基因组扫描图云平台支持从组装结果开始,共计13个模块多达30余项的分析统计内容。
平台支持客户自主上传数据进行分析,用户可在云平台结果展示页面查看各个模块分析结果,并对结果进行筛选、比对等分析,也可以对相应结果进行下载。
平台支持三种种类型的数据上传:
方式一:上传二代测序原始数据文件
基于原始数据,从组装开始,做完整套分析,分析数据最完整,强烈推荐使用这种方式。
方式二:上传细菌基因组扫描图组装结果
此方式适用于已经有组装结果的情况。流程基于上传的组装结果从基因预测模块开始执行并进行详细的各类注释分析。因为缺少二代测序数据,流程无法进行“测序数据质控与统计”、“基因组评估”(Kmer频率评估和GC-Depth分析)、“测序深度评估(分析各Scaffold的测序深度)”等板块的分析。
方式三:上传细菌基因组扫描图组装结果+二代测序数据
流程支持同时上传组装结果和二代测序数据,此方式不会对二代测序数据进行组装,可用于“测序数据质控与统计”、“基因组评估”(Kmer频率评估和GC-Depth分析)、“测序深度评估(分析各Scaffold的测序深度)”。流程基于上传的组装结果进行基因预测以及其他下游分析。整套分析数据完整,适用于自己单独优化组装或者特殊处理过的组装结果的情形。
详细操作步骤
Step1:注册登录(微信扫码+手机号验证)
网址:http://cloud.mimazi.net/login.html 或http://cloud2.mimazi.net:9001/login.html
首次使用请先点击上述网址进行注册,之后每次登录,只需要直接扫码登录即可。
Step2:选择云流程和设置参数
(1)初次使用,可以点击“7天试用”,可以免费使用7天;
(2)过期后,可以选择购买,分为月度型、半年型、全年型,在有效期内,均可直接使用;
(3)可以在“会员中心”--“项目中心”--“我的流程”,直接点击使用自己购买的流程,也可以直接在网站上直接选择“细菌基因组扫描图分析全套流程”,进行数据投递;
(4)填写参数信息,上传数据
建议先下载“示例”文件,查看详细的示例格式。
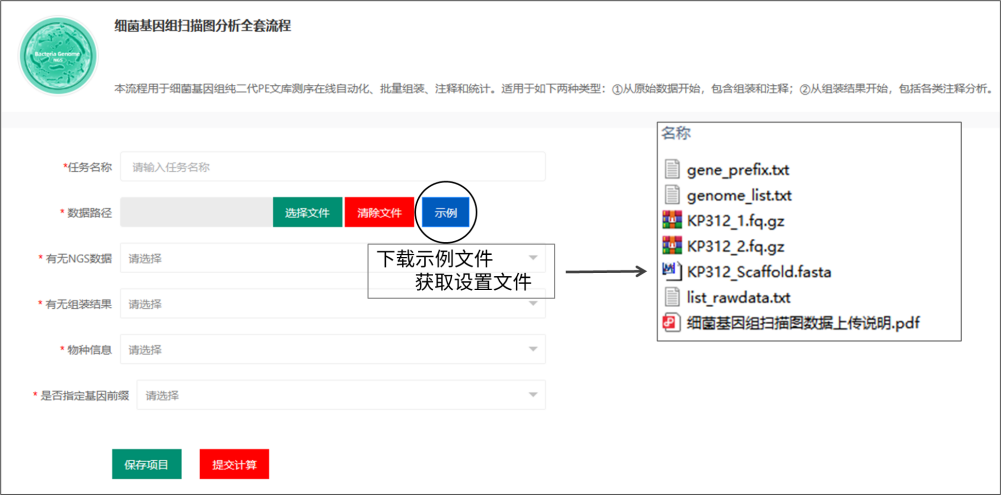
上传数据,建议使用http://cloud2.mimazi.net:9001网址登录,千兆网速。
A. 任务名称:自定义填写。建议填写可识别的项目名称,以便于自己可以精准地辨识出该任务具体分析的是什么样品;
B. 数据路径:选择存有数据的文件夹(不是具体的单个文件,是包含了所有待分析数据的文件夹!!!);
C. 有无NGS数据:如果没有二代测序数据,就选no,选择yes就要同时提供二代数据和“list_rawdata.txt”文件;
D. 是否校正组装:如果选择yes,需要同时提供二代数据,用于对组装结果的校正,如果选择no,可以提供也可以不提供二代测序数据,流程不会使用二代数据进行校正分析;
E. 物种信息:可根据实际情况选择,物种信息将用于ResFinder软件分析耐药基因,具体编号和物种对应关系如下所示:
SP0="Other"
SP1="Campylobacter jejuni"
SP2="Campylobacter coli"
SP3="Campylobacter spp."
SP4="Enterococcus faecalis"
SP5="Enterococcus faecium"
SP6="Escherichia coli"
SP7="Helicobacter pylori"
SP8="Klebsiella"
SP9="Mycobacterium tuberculosis"
SP10="Neisseria gonorrhoeae"
SP11="Plasmodium falciparum"
SP12="Salmonella spp."
SP13="Staphylococcus aureus"
F. 是否指定基因前缀:分析流程中,基因前缀默认为“gene”,若要指定,就选择“yes”,同时提供“gene_prefix.txt”文件。


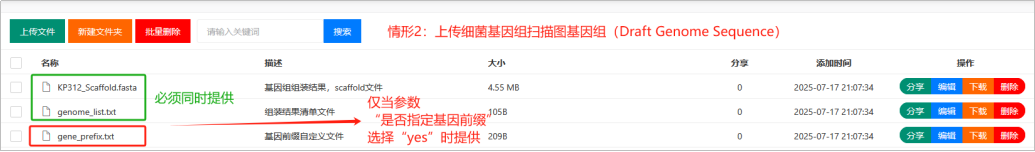
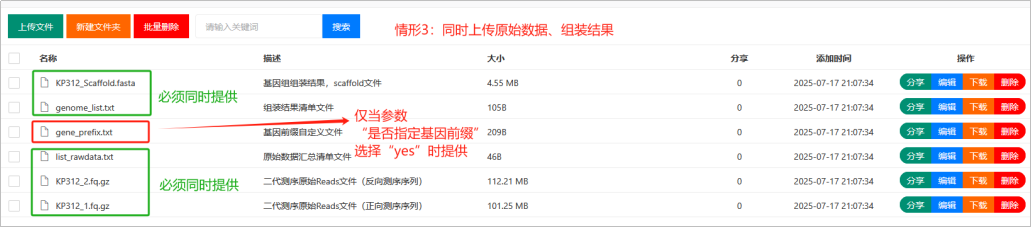

Step3:文件准备和格式确认
(1)二代NGS数据文件
流程支持从原始数据开始组装,之后进行基因预测和全套注释分析,此时,需要提供二代NGS数据文件。二代NGS数据文件,主要用于“测序数据质控与统计”、“基因组评估”(Kmer频率评估和GC-Depth分析)、“基因组组装”(若提供了组装结果文件,则流程不再使用原始数据进行组装)、“测序深度评估(分析各条Scaffold片段的测序深度)”。上传的NGS数据文件,必须是双末端测序数据,支持PE150、PE250、PE300读长。数据文件后缀名为*.fq.gz或者*.fastq.gz。文件名中不能有空格、不能有特殊字符。
当参数设置中选择“有无NGS数据”为“yes”且提供了原始数据时,需同时提供一个名为“list_rawdata.txt”的文件,该文件一共三列,表头分别是“Sample_Name”、“Raw1”、“Raw2”,以Tab键隔开。
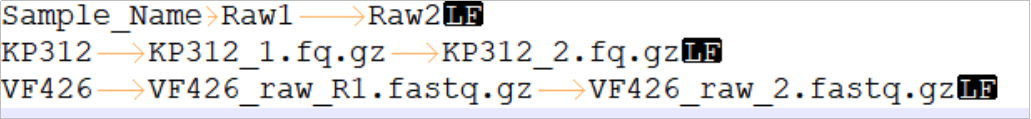
“list_rawdata.txt”文件的修改建议修改方式:
方式1:可以使用NotePad++软件(免费软件,网上可以自行下载安装)打开从示例文件中下载的“list_rawdata.txt”文件,然后把案例数据替换为自己的样本数据,保存后直接上传云计算平台;
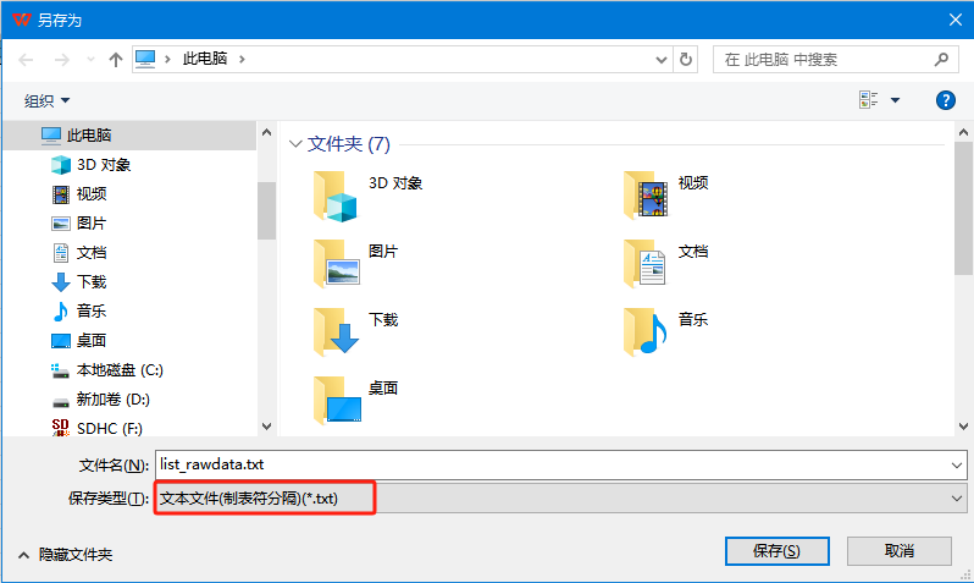
方式2:可以使用表格(如WPS、微软Excel)打开下载的“list_rawdata.txt”文件,替换和增添自己的样本数据以后,另存为“文本文件(制表符分割)(*.txt)”格式,上传云计算平台;
(2)组装结果文件
流程支持从组装结果开始,进行基因预测和下游的各类注释。上传的组装结果文件为标准的FastA格式文件,后缀名可以是*.fasta、*.fna、*.fa、*.fas、*.fnn等,例如KP312_Scaffold.fasta等,序列文件名称,不能包含空格及特殊字符。
若同时提供原始数据文件,则组装结果要求跟原始数据文件存储在同一个文件夹中。
(3)genome_list.txt文件
该文件名称和大小写都不能改变,跟组装结果文件存储在同一个文件夹中。
该文件共三列,以Tab键隔开,表头固定,不可更改,第一列,表头为“Sample_Name”,展示样本名称;第二列,表头为“File”,展示样本对应的组装序列,fasta格式,文件名称必须与实际上传的文件名称一致(包括后缀);第三列,表头为“Type”,标注为“Genome”,注意第一个字母大写。

编辑“genome_list.txt”文件内容的方式,参考“list_rawdata.txt”文件修改方式。
(4)基因前缀文件
基因前缀文件为非必须文件,流程中,默认基因前缀统一为“gene”,无需提供基因前缀相关的设置文件,此时基因ID规则为gene_0001、gene_0002……。流程支持自定义设置每个样品的基因前缀信息,若需自定义,可以在参数设置中“是否指定基因前缀”,选择“yes”,同时提供“gene_prefix.txt”文件。
该文件一共两列,表头分别是“Sample_Name”和“Gene_Prefix”,用Tab键隔开。原则上,“Gene_Prefix”可以自定义设置成各类字符,但建议跟自己样品的物种的缩写、菌株号关联起来,且如果后期上传NCBI,不能跟已公布的任何菌株的基因前缀雷同。
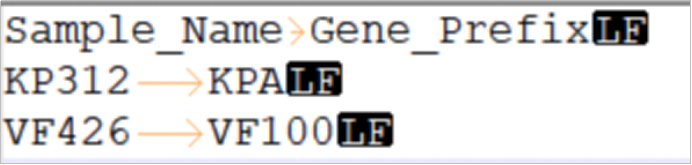
编辑“gene_prefix.txt”文件内容的方式,参考“list_rawdata.txt”文件修改方式。
Step4:分析运行与结果查看、结果下载
项目运行状态,可以在“会员中心”--“项目中心”--“云流程项目”栏目中查看,分为“运行中”、“已完成”、“运行失败”,相关log文件,可以在相应任务的“运行日志”中查看。
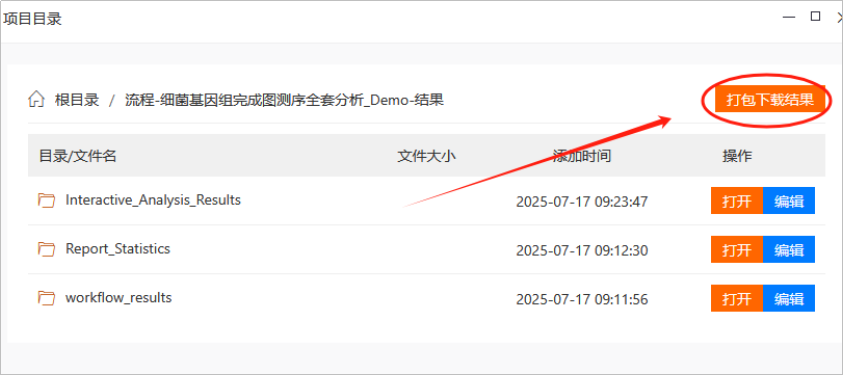
如果任务状态显示为“已完成”,可以点击任务名称,或者右侧的“结果”查看全部的结果数据,可以点击相应任务的“项目目录”,进入数据打包下载界面。
下载数据,建议使用http://cloud2.mimazi.net:9001/login.html网址登录,千兆网速。
重要提示
(1)“list_rawdata.txt”文件、“gene_prefix.txt”文件、“genome_list.txt”文件中的样本量、样本名,必须与中的样本信息一致,样本数量不能多,也不能少。
(2)单次上传样本量,最多为30个,如果样品较多,可以分批次分析。
(3)上传和下载数据,强烈建议使用http://cloud2.mimazi.net:9001网址登录,千兆网速。
(4)遇到实操问题,可以联系我们技术部客服(微信)协助解决。
当前版本为 1.0b版本,上架时间为2025年7月5日
- 暂无推荐课程