- 软件介绍
- 软件与数据库
- 使用指南
- 版本记录
- 相关课程推荐
细菌基因组完成图流程支持用户自主上传数据,针对上传数据进行全套基因组注释分析,用户可在结果展示页面对所有分析结果分模块进行查看,并可进行结果的筛选、排序、下载等操作。
平台支持两种数据上传方式:
第一种,上传完成图组装结果,包括质粒和染色体序列。针对上传的组装序列从基因预测开始执行全套分析。
第二种,上传完成图组装结果+二代测序原始数据。除基因组本身的分析之外,还可对二代数据进行质控统计,并基于二代数据进行基因组评估、测序深度计算等。
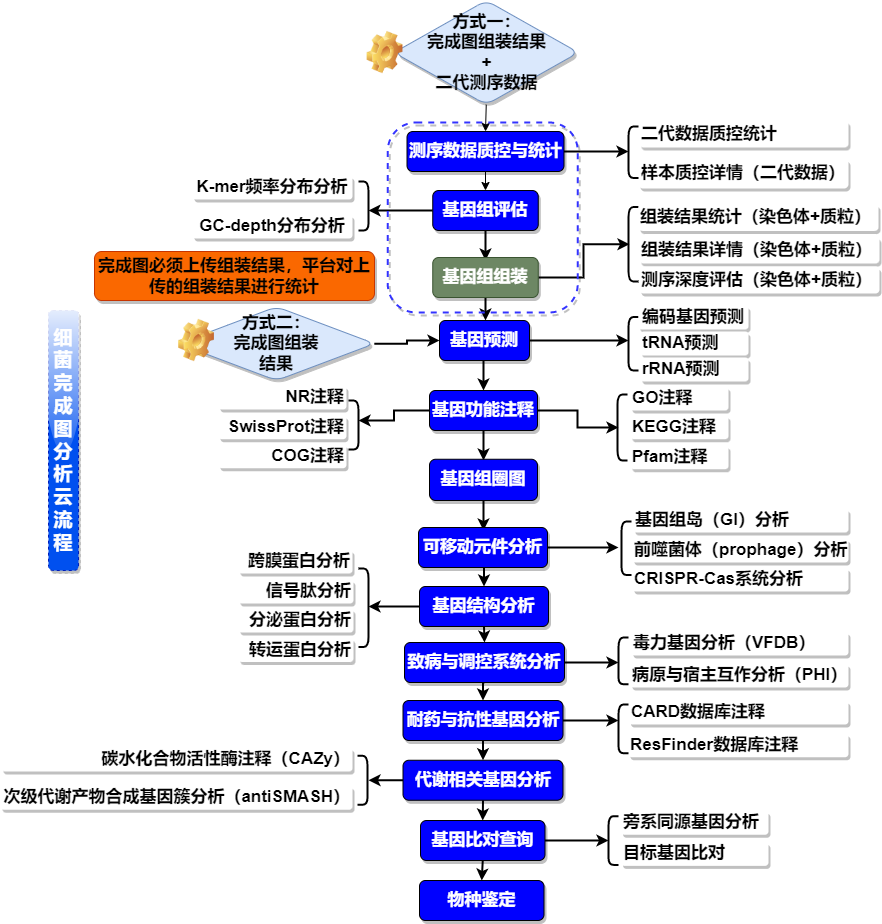
基因预测:
CDS预测:Glimmer(http://ccb.jhu.edu/software/glimmer/index.shtml,V3.02)
tRNA预测:tRNAscan-SE(http://trna.ucsc.edu/software/)
rRNA预测:Barrnap(https://github.com/tseemann/barrnap/)
基因功能注释:
比对软件:DIOMAND(V0.9.24.125,http://github.com/bbuchfink/diamond)
比对数据库:
NR:NCBI创建并维护的非冗余蛋白数据库,数据库内容较为全面,可作为基因注释结果的主要参考。
Swiss-Prot(https://web.expasy.org/docs/swiss-prot_guideline.html):由欧洲生物信息学研究所(European Bioinformatics Institute,EBI)维护的非冗余基因数据库,注释结果大多经过实验验证,可靠性较高,但收录基因数量很少。
Pfam(http://pfam.xfam.org/):依赖于由多序列比对和隐马尔可夫模型(HMMs),可给出基因中包含的所有结构域信息。
COG(http://www.ncbi.nlm.nih.gov/COG/):Clusters of Orthologous Groups of Proteins的缩写,可以对预测蛋白进行功能注释、归类以及蛋白进化分析。构成一个COG对应于一个古老的保守结构域,每个COG的蛋白被假定来自于同一个祖先蛋白。
GO(http://www.geneontology.org/):基因本体论Gene Ontology的缩写,为标准化不同物种、不同数据库的生物学术语而创立的数据库,可获得标准化的功能描述信息。
KEGG(http://www.genome.jp/kegg/):是系统分析基因功能,联系基因组和功能信息的大型知识库,其丰富的通路信息有助于从系统水平去了解基因的生物学功能,例如代谢途径、遗传信息传递以及细胞学过程等一些复杂的生物过程。
基因组圈图绘制:Circos Version 0.69-6(http://www.circos.ca)
特定数据库注释:
基因组岛:IslandViewer 4
前噬菌体:PHASTER
CRISPR-Cas系统:CRISPRFinder
碳水化合物活性酶:CAZy(http://www.cazy.org/)
毒力基因:VFDB
耐药基因:CARD&ResFinder
病原菌与宿主互作:PHI(http://www.phi-base.org/)
跨膜蛋白:TMHMM
转运蛋白:TCDB
分泌蛋白:SignalP(http://www.cbs.dtu.dk/services/SignalP)
次级代谢产物合成基因簇:antiSMASH
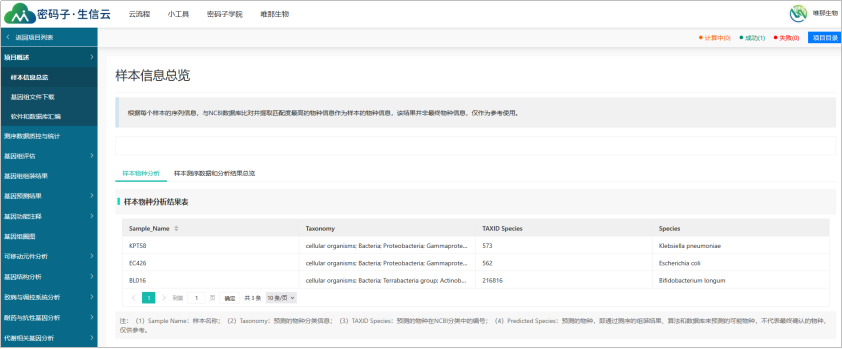
细菌基因组完成图云平台支持从组装结果开始,共计13个模块多达30余项的分析统计内容。
平台支持客户自主上传数据进行分析,用户可在云平台结果展示页面查看各个模块分析结果,并对结果进行筛选、比对等分析,也可以对相应结果进行下载。
完成图基因组组装涉及完整性判断、基因组起始位点判断等工作,都是基于比较精细的人工判断,软件直接组装的结果往往不能直接使用,所以完成图云分析流程必须提供已经组装判断好的基因组组装序列。
平台支持两种种类型的数据上传:
方式一:仅上传细菌完成图组装结果
流程基于上传的组装结果从基因预测模块开始执行。因为没有原始数据,因此与原始数据相关模块的结果缺失,包括“测序数据质控与统计”、“基因组评估”(Kmer频率评估和GC-Depth分析)、“测序深度评估(分析染色体、质粒的测序深度)”。
方式二:上传完成图组装结果+二代测序数据
流程支持同时上传组装结果和二代测序数据,此方式不会对二代测序数据进行组装,可用于“测序数据质控与统计”、“基因组评估”(Kmer频率评估和GC-Depth分析)、“测序深度评估(分析染色体、质粒的测序深度)”、“对三代组装结果进行校正”(需选择校正为yes)。流程基于上传的组装结果(或校正后的组装结果)进行基因预测以及其他下游分析。
详细操作步骤
Step1:注册登录(微信扫码+手机号验证)
网址:http://cloud.mimazi.net/login.html 或http://cloud2.mimazi.net:9001/login.html
首次使用请先点击上述网址进行注册,之后每次登录,只需要直接扫码登录即可。
Step2:选择云流程和设置参数
(1)初次使用,可以点击“7天试用”,可以免费使用7天;
(2)过期后,可以选择购买,分为月度型、半年型、全年型,在有效期内,均可直接使用;
(3)可以在“会员中心”--“项目中心”--“我的流程”,直接点击使用自己购买的流程,也可以直接在网站上直接选择“细菌基因组完成图分析全套流程”,进行数据投递;

(4)填写参数信息,上传数据
建议先下载“示例”文件,查看详细的示例格式。
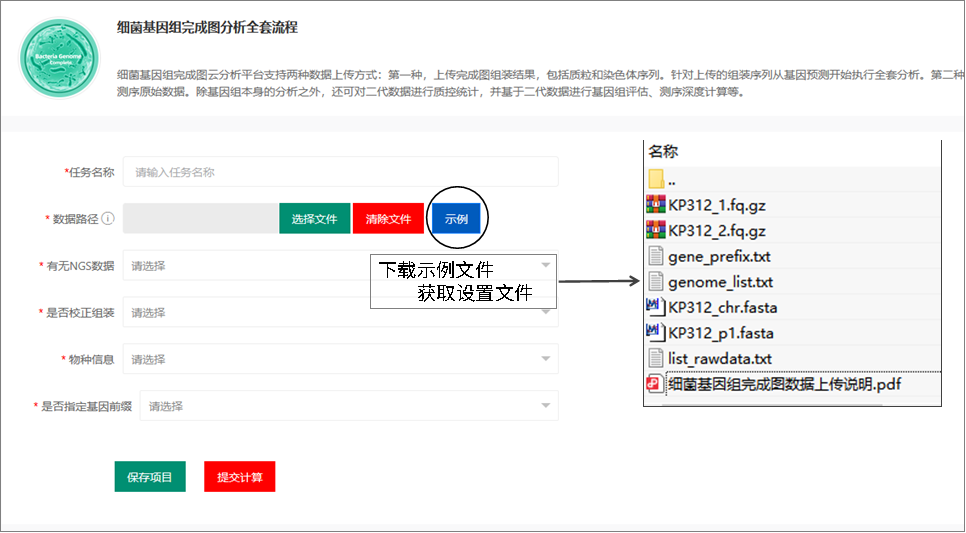
上传数据,建议使用http://cloud2.mimazi.net:9001网址登录,千兆网速。
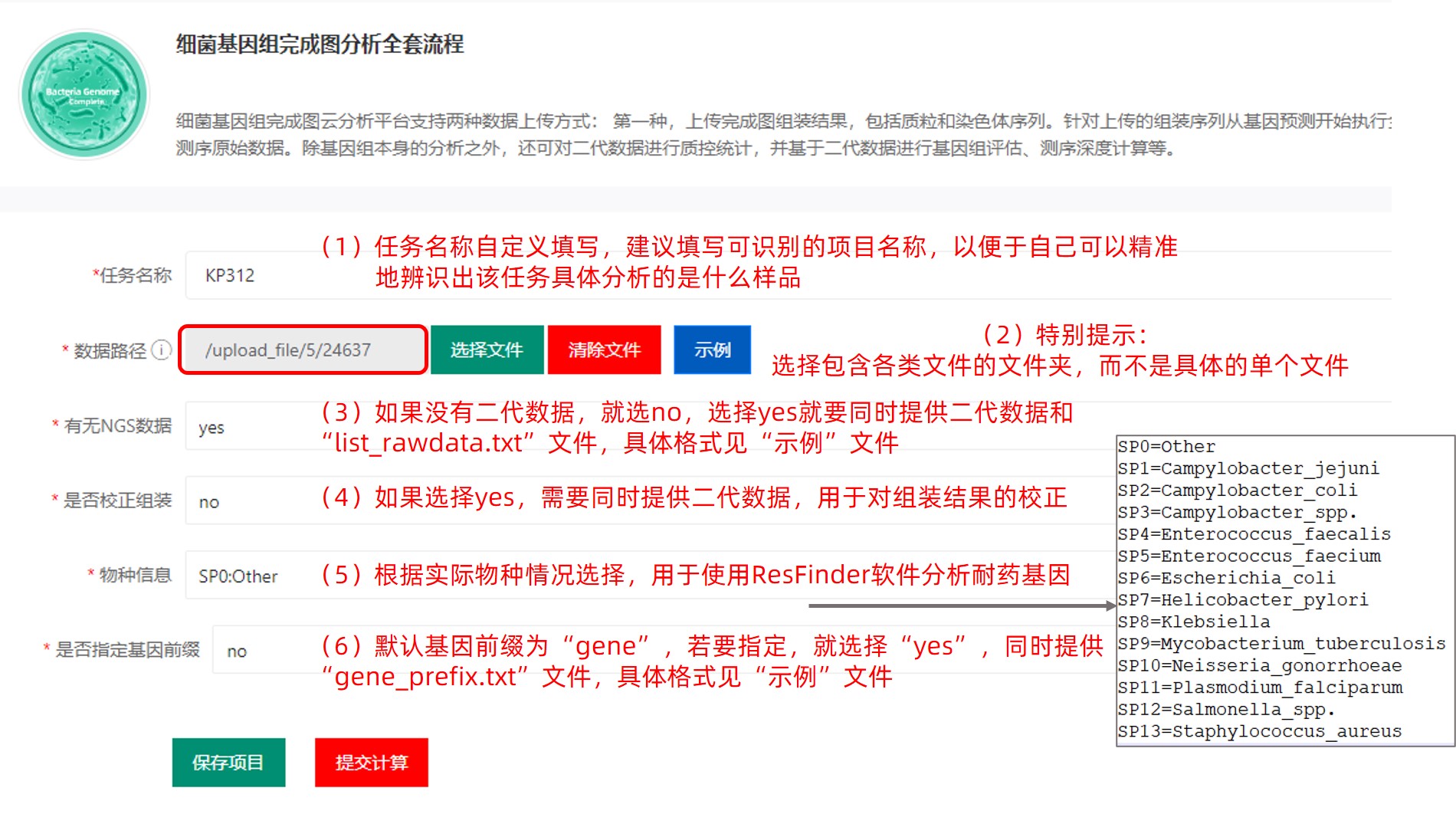
A. 任务名称:自定义填写。建议填写可识别的项目名称,以便于自己可以精准地辨识出该任务具体分析的是什么样品;
B. 数据路径:选择存有数据的文件夹(不是具体的单个文件,是包含了所有待分析数据的文件夹!!!);
C. 有无NGS数据:如果没有二代测序数据,就选no,选择yes就要同时提供二代数据和“list_rawdata.txt”文件;
D. 是否校正组装:如果选择yes,需要同时提供二代数据,用于对组装结果的校正,如果选择no,可以提供也可以不提供二代测序数据,流程不会使用二代数据进行校正分析;
E. 物种信息:可根据实际情况选择,物种信息将用于ResFinder软件分析耐药基因,具体编号和物种对应关系如下所示:
SP0="Other"
SP1="Campylobacter jejuni"
SP2="Campylobacter coli"
SP3="Campylobacter spp."
SP4="Enterococcus faecalis"
SP5="Enterococcus faecium"
SP6="Escherichia coli"
SP7="Helicobacter pylori"
SP8="Klebsiella"
SP9="Mycobacterium tuberculosis"
SP10="Neisseria gonorrhoeae"
SP11="Plasmodium falciparum"
SP12="Salmonella spp."
SP13="Staphylococcus aureus"
F. 是否指定基因前缀:分析流程中,基因前缀默认为“gene”,若要指定,就选择“yes”,同时提供“gene_prefix.txt”文件。


Step3:文件准备和格式确认
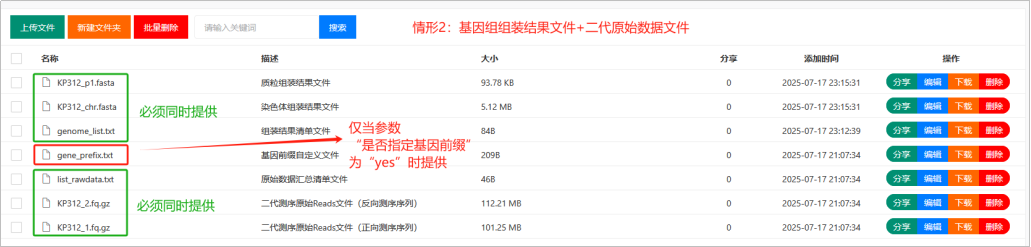
(1)组装结果文件(必须文件)
要求细菌基因组完成图的组装结果文件为标准的FastA格式文件,后缀名可以是*.fasta、*.fna、*.fa、*.fas、*.fnn等,如果样本中包含有多个复制单元,比如两条染色体、一个或多个质粒等等,则需要将不同复制单元单独保存,不能混合在一个FastA文件中,序列文件名称,不能包含空格及特殊字符。例如KP312_chr.fasta、KP312_p1.fasta、KP312_p2.fasta等。
组装结果存储于特定文件夹中。
(2)genome_list.txt文件(必须文件)
该文件名称不能改变,跟组装结果文件存储在同一个文件夹中。
该文件共三列,表头固定,不可更改,第一列,表头为“Sample_Name”,展示样本名称;第二列,表头为“File”,展示样本对应的每个复制子的组装序列,fasta格式,文件名称必须与实际上传的文件名称一致(包括后缀);第三列,表头为“Type”,即复制子的类型,染色体标注“Chromosome”,质粒标注“Plasmid”,注意这两个字段是固定的,不可修改,注意第一个字母大写。

修改建议修改方式:
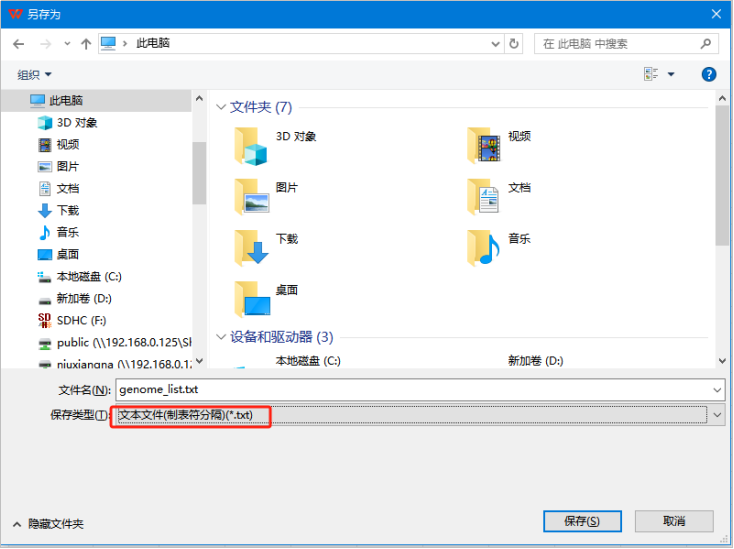
方式1:可以使用NotePad++软件打开genome_list.txt文件,然后把案例数据替换为自己的样本数据,直接保存后上传云计算平台;
方式2:可以使用表格(如WPS、微软Excel)打开genome_list.txt文件,替换和增添自己的样本数据以后,另存为“文本文件(制表符分割)(*.txt)”格式,上传云计算平台。
(3)基因前缀文件(非必须文件)
基因前缀文件为非必须文件,流程中,默认基因前缀统一为“gene”,无需提供基因前缀相关的设置文件,此时基因ID规则为gene_0001、gene_0002……。流程支持自定义设置每个样品的基因前缀信息,若需自定义,可以在参数设置中“是否指定基因前缀”,选择“yes”,同时提供“gene_prefix.txt”文件。
该文件一共两列,表头分别是“Sample_Name”和“Gene_Prefix”,用tab键隔开。原则上,“Gene_Prefix”可以自定义设置成各类字符,但建议跟自己样品的物种的缩写、菌株号关联起来,且如果后期上传NCBI,不能跟已公布的任何菌株的基因前缀雷同。

编辑“gene_prefix.txt”文件内容的方式,参考genome_list.txt文件修改方式。
(4)二代NGS数据文件(非必须文件)
流程支持同时上传二代NGS数据文件,主要用于“测序数据质控与统计”、“基因组评估”(Kmer频率评估和GC-Depth分析)、“测序深度评估(分析染色体、质粒的测序深度)”、“对三代组装结果进行校正”(需选择校正为yes)。上传的NGS数据文件,必须是双末端测序数据,支持PE150、PE250、PE300读长。数据文件后缀名为*.fq.gz或者*.fastq.gz。文件名中不能有空格、不能有特殊字符。
NGS数据,要求跟组装结果文件存储在同一个文件夹中。
当参数设置中选择“有无NGS数据”为“yes”且提供了原始数据时,需同时提供一个名为“list_rawdata.txt”的文件,该文件一共三列,表头分别是“Sample_Name”、“Raw1”、“Raw2”,以Tab键隔开。

编辑“list_rawdata.txt”文件内容的方式,参考genome_list.txt文件修改方式。
Step4:分析运行与结果查看、结果下载
项目运行状态,可以在“会员中心”--“项目中心”--“云流程项目”栏目中查看,分为“运行中”、“已完成”、“运行失败”,相关log文件,可以在相应任务的“运行日志”中查看。
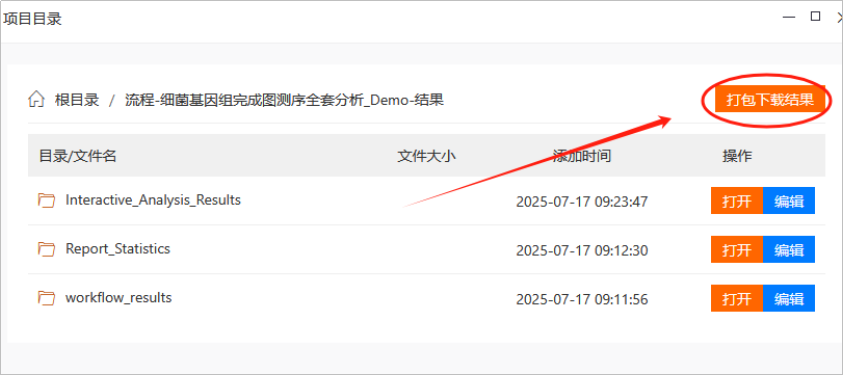
如果任务状态显示为“已完成”,可以点击任务名称,或者右侧的“结果”查看全部的结果数据,可以点击相应任务的“项目目录”,进入数据打包下载界面。
下载数据,建议使用http://cloud2.mimazi.net:9001/login.html网址登录,千兆网速。
重要提示
(1)如果提供“list_rawdata.txt”文件、“gene_prefix.txt”文件,则文件中的样本量、样本名,必须与“genome_list.txt”文件中的样本信息一致,样本数量不能多,也不能少。
(2)单次上传样本量,最多为30个,如果样品较多,可以分批次分析。
(3)上传和下载数据,强烈建议使用http://cloud2.mimazi.net:9001/login.html网址登录,千兆网速。
(4)遇到实操问题,可以联系我们技术部客服(微信)协助解决。
本流程为Version 1.0版本,上线时间为2025年7月10日。
- 暂无推荐课程